
附属眼科医院李柯然、姚进课题组发现脑源性神经营养因子信号增强子Syn3对糖尿病视网膜病变神经损伤的保护作用
5月15日,附属眼科医院李柯然副教授、姚进教授课题组,苏州大学曹聪教授课题组联合在《蛋白质和细胞(Protein & Cell)(高水平4类,Q1,IF = 21.2)发表了题为《Syn3,一种新开发的环肽和BDNF信号增强子,改善糖尿病视网膜病变的视网膜神经节细胞变性》(Syn3, a newly developed cyclic peptide and BDNF signaling enhancer, ameliorates retinal ganglion cell degeneration in diabetic retinopathy)研究论文。该研究揭示了Syn3通过强化BDNF信号显著抑制DR的神经损伤。李柯然和其课题组的硕士研究生李佳骏、姚进教授课题组的硕士研究生曹原为本文的共同第一作者。
糖尿病视网膜病变(diabetic retinopathy, DR)是全球范围内面临的巨大公共卫生挑战,也是全球致盲的主要原因之一。视网膜神经-血管单元(NVU)由神经元、血管内皮细胞、胶质细胞、周细胞、基底膜及细胞外基质等组分共同构成,其通过复杂的动态交互网络共同维持视网膜内环境稳态。神经元作为NVU的核心组分,其损伤与凋亡不仅会直接影响视觉信号的传递与处理,更会破坏NVU各组分间的动态平衡,进而触发血管内皮细胞功能障碍、胶质细胞过度活化及炎症反应等病理过程,深入探索有效的神经保护策略以维持NVU稳态,对于预防DR的发生发展,保护DR患者的视功能具有重要意义。
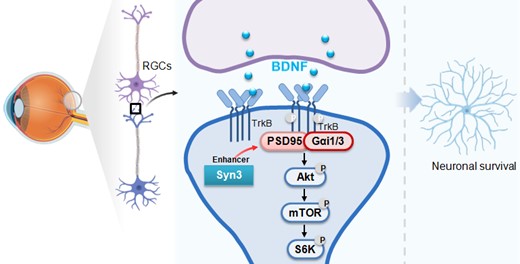
脑源性神经营养因子(Brain Derived Neurotrophic Factor,BDNF)是中枢神经系统中最为经典且最具特征的神经营养因子,BDNF与其受体酪氨酸激酶B(TrkB)结合可活化下游信号通路,发挥神经保护及增强突触强度等多重作用。课题组前期合作研发了BDNF信号enhancer(增强子)--突触后致密蛋白95(PSD95)结合肽CN2097。CN2097结合PSD95的PDZ3结构域后易化BDNF诱导的TrkB-PSD95-Gαi1/3耦联及下游Akt-mTOR及CaMKII的活化,促神经元存活。但CN2097和PSD95的PDZ3结构域结合的亲和力相对较低,所需要的有效浓度相对较高。在此基础上,进一步将CN2097氨基酸序列进行进一步的结构优化并筛选了一种新的极高效的环肽化合物:命名为“Syn3”。有别于CN2097,Syn3与PDZ3的扩展的αC螺旋结合,它和PSD95的PDZ3结构域结合的亲和力比CN2097高10倍左右,且更易透过血-眼屏障。
研究发现,在原代培养的RGCs中,添加200 nM(CN2097有效浓度的十分之一)的Syn3显著强化BDNF(25 ng/mL)诱导的TrkB-Gαi1/3-PSD95互作及下游Akt-S6K的活化。但Syn3不改变TrkB的表达、磷酸化及Gαi1/3-PSD95的表达。RGCs中慢病毒shRNA方法敲减TrkB、PSD95或Gαi1/3后阻断Syn3强化BDNF信号的作用。体外氧葡萄糖剥夺/再氧化(OGD/R)模型诱导了RGCs损伤和凋亡;而Syn3联合BDNF给药显著抑制了OGD/R诱导的RGCs凋亡,并促细胞存活。
在体动物实验发现玻璃体腔内注射Syn3可显著抑制DR模型鼠的RGCs丢失。运用玻璃体腔内注射腺病毒的方法,小鼠神经元特异性敲减Gαi1/3后显著抑制了Akt-mTOR的激活,并减少了视网膜神经节细胞层(GCL)中RGCs的数量。此时添加Syn3未增加Gαi1/3敲减小鼠中的Akt-mTOR活化及RGCs的数量。反之,DR模型小鼠中神经元特异性过表达Gαi1/3后增强了Akt-mTOR的激活,并缓解了GCL中RGCs损伤。
该研究明确了神经元功能障碍是DR发生发展及NVU稳态失衡中的关键事件,创新性地研发了针对糖尿病性视网膜病变神经损伤的靶向保护药物,从NVU的角度,寻找DR的前哨机制和干预策略,有望为DR等致盲性眼病的早防早治提供新的干预手段和防治策略。
原文链接:
https://academic.oup.com/proteincell/advance-article/doi/10.1093/procel/pwae028/7671574?login=true#